____________________________________________ 5
Biochemistry
5.1 DNA
5.1.1 The double helix
The sentence "This structure has novel features which are of considerable biological interest" may be one of science's most famous understatements. It appeared in April 1953 in the scientific paper where James Watson and Francis Crick presented the structure of the DNA-helix, the molecule that carries genetic information from one generation to the other.
Nine years later, in 1962, they shared the Nobel Prize in Physiology or Medicine with Maurice Wilkins, for solving one of the most important of all biological riddles. Half a century later, important new implications of this contribution to science are still coming to light. (Fredholm, n.d.)
The three-dimensional structure of DNA – The double helix- arises from the chemical and structural features of its two polynucleotide chains. Because these two chains are held together by hydrogen bonding between the bases on the different strands, all the bases are on the inside of the double helix, and the sugar -phosphate backbones are on the outside. In each case, a bulkier two-ring base (a purine) is paired with a single-ring base (a pyrimidine); Adenine always pairs with Thymine, and Guanine with Cytosine. The complementary base-pairing enables the base pairs to be packed in the energetically most favorable arrangement in the interior of the double helix. In this arrangement, each base pair is of similar width, thus holding the sugar-phosphate backbones an equal distance apart along the DNA molecule. To maximize the efficiency of base-pair packing, the two sugar-phosphate backbones wind around each other to form a double helix, with one complete turn every ten base pairs. (Alberts, et al., 2002)
5.1.2 Protein synthesis
DNA stores the information for protein synthesis and RNA carries out the instructions encoded in DNA; most biological activities are carried out by proteins. The accurate synthesis of proteins thus is critical to the proper functioning of cells and organisms. The linear order of amino acids in each protein determines its three-dimensional structure and activity. For this reason, assembly of amino acids in their correct order, as encoded in DNA, is the key to production of functional proteins. (Lodish, et al., The Three Roles of RNA in Protein Synthesis, 2000)
Although it is universally accepted that protein synthesis occurs in the cytoplasm, the possibility that translation can also take place in the nucleus has been hotly debated. Reports have been published claiming to demonstrate nuclear translation, but alternative explanations for these results have not been excluded, and other experiments argue against it. Much of the appeal of nuclear translation is that functional proofreading of newly made mRNAs in the nucleus would provide an efficient way to monitor mRNAs for the presence of premature termination codons, thereby avoiding the synthesis of deleterious proteins. mRNAs that are still in the nucleus-associated fraction of cells are subject to translational proofreading resulting in nonsense-mediated mRNA decay and perhaps nonsense-associated alternate splicing. However, these mRNAs are likely to be in the perinuclear cytoplasm rather than within the nucleus. Therefore, in the absence of additional evidence, nuclear translation is unlikely to occur. (Dahlberg & Lund, 2004) (Dahlberg & Lund, 2004)
5.2 Genomics
5.2.1 DNA sequencing
Recent scientific discoveries that resulted from the application of next generation DNA sequencing technologies highlight the striking impact of these massively parallel platforms on genetics. These new methods have expanded previously focused readouts from a variety of DNA preparation protocols to a genome-wide scale and have fine-tuned their resolution to single base precision. The sequencing of RNA also has transitioned and now includes full-length cDNA analyses, serial analysis of gene expression (SAGE)-based methods, and noncoding RNA discovery. Next-generation sequencing has also enabled novel applications such as the sequencing of ancient DNA samples and has substantially widened the scope of metagenomic analysis of environmentally derived samples. Taken together, an astounding potential exists for these technologies to bring enormous change in genetic and biological research and to enhance our fundamental biological knowledge. (Mardis, 2008)
5.2.3 Comparative genomics
Comparative genomics is a field of biological research in which the genome sequences of different species — human, mouse, and a wide variety of other organisms from bacteria to chimpanzees — are compared. By comparing the sequences of genomes of different organisms, researchers can understand what, at the molecular level, distinguishes different life forms from each other. Comparative genomics also provides a powerful tool for studying evolutionary changes among organisms, helping to identify genes that are conserved or common among species, as well as genes that give each organism its unique characteristics.
Comparison of discrete segments of genomes is also possible by aligning homologous DNA from different species. As an example, a human gene (pyruvate kinase: PKLR) and the corresponding PKLR homologs from macaque, dog, mouse, chicken, and zebrafish are aligned. Regions of high DNA sequence similarity with human across a 12-kilobase region of the PKLR gene are plotted for each organism. A high degree of sequence similarity between human and macaque (two primates) in both PKLR exons (blue) as well as introns (red) and untranslated regions (light blue) of the gene. In contrast, the chicken and zebrafish alignments with human only show similarity to sequences in the coding exons; the rest of the sequence has diverged to a point where it can no longer be reliably aligned with the human DNA sequence. Using such computer-based analysis to zero in on the genomic features that have been preserved in multiple organisms over millions of years, researchers are able to locate the signals that represent the location of genes, as well as sequences that may regulate gene expression. Indeed, much of the functional parts of the human genome have been discovered or verified by this type of sequence comparison and it is now a standard component of the analysis of every new genome sequence. (Touchman, 2010)
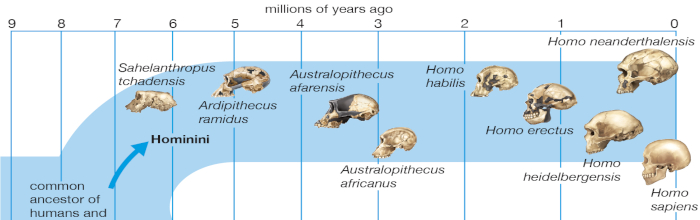
5.3 Paleoproteomics
5.3.1 Ancient proteins
Although the findings were controversial—some researchers still doubt that proteins can resist degradation for tens of millions of years— A small but growing number of researchers specializing in the analysis of ancient proteins, or paleoproteomics, to learn about the biology of organism’s past. It’s been a goal of scientists for some time now; in the 1950s, several researchers were already discussing the possibility of studying peptides preserved in fossils. But only in the last two decades have advances in techniques for protein analysis, such as mass spectrometry, made the feat practical.
The potential for learning about ancient life from paleoproteomics is substantial. Via their amino acid sequences, peptides offer many of the same insights as DNA about genomic makeup—information that can support new or existing phylogenetic trees, inform research on past migrations, and assist with species identifications, even amidst a jumble of ancient remains. But proteins tend to last longer in the geological record than nucleic acids, thanks to both greater volumes at deposition and more-degradation-proof molecular structures. “Both DNA and proteins are chains of building blocks,” explains Cappelini, a paleoproteomics researcher at the Natural History Museum of Denmark. “But the bonds connecting those blocks are more stable in proteins than in DNA.” The oldest confirmed DNA samples, extracted from ice cores taken in southern Greenland, are less than 800,000 years old, while the oldest protein, even by conservative estimates, dates back several million years. (Offord, 2018)
5.3.2 A novel sequencing approach
The challenge with older fossil DNA sequencing is that the progressive fragmentation of DNA even under optimal conditions, a function of time and temperature, results in ever shorter fragments of DNA. This process continues until no DNA can be sequenced or reliably aligned. Ancient proteins ultimately suffer a similar fate but are a potential alternative source of biomolecular sequence data to investigate hominin phylogeny given their slower rate of fragmentation. In addition, ancient proteins have been proposed to potentially provide insights into in vivo biological processes and can be used to provide additional ecological information through large scale ZooMS (Zooarchaeology by Mass Spectrometry) screening of unidentifiable bone fragments. (Welker, 2018)
5.4 Human Biochemistry
5.4.1 Sequencing the human genome
It is apparent that molecular biology formed a direct link between the genes located on the chromosomes and the sequence data each gene carries. Fragments of DNA have been snipped off along each chromosome to allow “shotgun sequencing” developed in deciphering the human genome, where bacterial vectors are used to clone random fragments of a long DNA molecule. Fragments are then sequenced in parallel and reads are assembled using their overlaps. Polymerase chain reaction is an earlier method developed and still in use.
In the 1980s, the advent of information technology laid the foundations of bioinformatics. Processor computing allowed to automate the general principles of overlapping sequences by similarity using dedicated computer programs. More recent developments are nano technologies that rely on the detection of an electrical rather than an optical signal. Nanopore devices can be as small as a USB stick. In 2016, such portability allowed to sequence Ebola virus at field sites in West Africa in less than 60 min. (Giani, Gallo, Gianfranceschi, & Formenti, 2020)
Research methods developed during the sequencing of the human genome opened the door into a broader area of inquiry. Genomic data has made possible the construction of evolutionary lineages and human biological variation. The foremost applications of molecular anthropology have been in unraveling the evolutionary relationship of extant species to sketch phylogenetic trees that will lead at the distant common ancestor. The approximate date of divergence of groups can also be estimated as has been achieved with the comparative study of the universal protein albumin.
Biomedical research has benefited from genomic data where human diseases that have a genetic basis
have been identified. The current practice of preventative medicine through genomic diagnosis maybe an expensive, but certainly, a promising approach.
The US Department of Energy has taken an active part in genomic research. To harness this potential, the U.S. Department of Energy’s Genomic Science program supports fundamental research to understand the systems biology of plants and microbes as they respond to and modify their environment.
5.4.2 Variations in human genomics
Emerging technologies now make it possible to genotype hundreds of thousands of genetic variations in individuals, across the genome. The study of loci at finer scales will facilitate the understanding of genetic variation at genomic and geographic levels. Global and chromosomal variations were examined across HapMap populations using 3.7 million single nucleotide polymorphisms to search for the most stratified genomic regions of human populations and linked these regions to ontological annotation and functional network analysis. To achieve this, five complementary statistical and genetic network procedures were used: principal component (PC), cluster, discriminant, fixation index (FST) and network/pathway analyses. At the global level, the first two PC scores were sufficient to account for major population structure; however, chromosomal level analysis detected subtle forms of population structure within continental populations, and as many as 31 PCs were required to classify individuals into homogeneous groups. Using recommended population ancestry differentiation measures, a total of 126 regions of the genome were catalogued. Gene ontology and networks analyses revealed that these regions included the genes encoding oculocutaneous albinism II (OCA2), hect domain and RLD 2 (HERC2), ectodysplasin A receptor (EDAR) and solute carrier family 45, member 2 (SLC45A2). These genes are associated with melanin production, which is involved in the development of skin and hair color, skin cancer and eye pigmentation. We also identified the genes encoding interferon-γ (IFNG) and death-associated protein kinase 1 (DAPK1), which are associated with cell death, inflammatory and immunological diseases. An in-depth understanding of these genomic regions may help to explain variations in adaptation to different environments. Our approach offers a comprehensive strategy for analyzing chromosome-based population structure and differentiation and demonstrates the application of complementary statistical and functional network analysis in human genetic variation studies. (Baye, 2011)
Human endogenous retroviruses (HERV) sequences account for about 8% of the human genome. Through comparative genomics and literature mining, a total of 29 human-specific HERV-K insertions were identified and characterized focusing on their structure and flanking sequence. The results showed that four of the human-specific HERV-K insertions deleted human genomic sequences via non-classical insertion mechanisms. Interestingly, two of the human-specific HERV-K insertion loci contained two HERV-K internals and three LTR elements, a pattern which could be explained by LTR-LTR ectopic recombination or template switching. In addition, a polymorphic test shows that twelve out of the 29 elements are polymorphic in the human population. In conclusion, human-specific HERV-K elements have inserted into human genome since the divergence of human and chimpanzee, causing human genomic changes. Thus, it is believed that human-specific HERV-K activity has contributed to the genomic divergence between humans and chimpanzees, as well as within the human population. (Shin, et al., 2013)
5.4.3 Applications
Unlocking the information contained within the human genome will likely advance our understanding of cardiovascular (CV) health and disease by leading to discovery of new molecules, pathways, and networks. A central strategy in genetic studies of CV disease has been to correlate human genomic DNA variation with clinical phenotypes, such as myocardial infarction, heart failure, stroke, and their risk factors, with a range of experimental designs and analytical procedures. The ability to detect genomic differences between individuals is the foundation of this research. Human genomic variation exists in many forms, each of which has unique qualitative and quantitative features. Each form of human genomic variation is composed of many individual variants that occur across the genome. The population frequency of individual variants can range from rare to common. The effect of a specific genomic variant can range from beneficial to knowledge into diagnosis and treatment of CV disease, it is logical to search for common genomic variants that have a non-neutral impact. In the recent past, one form of genomic variation, the single-nucleotide variant, has dominated the experimental landscape: It is the currency of present genetic CV disease studies. However, recent developments indicate that the focus on single-nucleotide polymorphisms (SNPs) alone will not capture the full range of meaningful human genomic variation, such as a newly characterized and annotated form called copy number variation (CNV). The main forms of human genomic variation are shown in Figure 1. These include SNPs, which are qualitative in nature and involve only a single nucleotide, and a family of genomic changes collectively called structural variations, which are quantitative in nature because they affect the dosage or copy number of a particular genomic region Structural variant types include deletions, duplications, inversions, and rearrangements of “chunks” of the genome. (Pollex & Hegele, 2007)
Fostering data sharing is a scientific and ethical imperative. Health gains can be achieved more comprehensively and quickly by combining large, information-rich datasets from across conventionally siloed disciplines and geographic areas. While collaboration for data sharing is increasingly embraced by policymakers and the international biomedical community, we lack a common ethical and legal framework to connect regulators, funders, consortia, and research projects so as to facilitate genomic and clinical data linkage, global science collaboration, and responsible research conduct. Governance tools can be used to responsibly steer the sharing of data for proper stewardship of research discovery, genomics research resources, and their clinical applications. It has been proposed that an international code of conduct be designed to enable global genomic and clinical data sharing for biomedical research. To give this proposed code universal application and accountability, it will be positioned within a human rights framework. This proposition is not without precedent: international treaties have long recognized that everyone has a right to the benefits of scientific progress and its applications, and a right to the protection of the moral and material interests resulting from scientific productions. It is time to apply these twin rights to internationally collaborative genomic and clinical data sharing. (Knoppers, Harris, Budin-Ljøsne, & Dove, 2014)